Regulatory Affairs Professionals
Regulatory Affairs Professionals play a critical role in industries such as pharmaceuticals, biotechnology, medical devices, and other sectors that are highly regulated. Their primary responsibility is to ensure that companies comply with all the regulatory requirements set forth by governmental agencies and regulatory bodies such as the FDA (Food and Drug Administration) in the U.S., EMA (European Medicines Agency) in Europe, or similar agencies in other regions. Here’s a breakdown of their key functions and responsibilities:
- Regulatory Compliance: They ensure that products (such as drugs, medical devices, or biologics) comply with all regulatory laws, guidelines, and requirements at every stage of development, from research and development to marketing and post-marketing surveillance.
- Documentation and Submissions: Regulatory Affairs (RA) professionals prepare and submit dossiers, including new drug applications (NDAs), investigational new drug applications (INDs), and marketing authorizations, to obtain approval for product commercialization.
- Liaison with Regulatory Agencies: They act as a bridge between the company and regulatory authorities, addressing any questions, providing necessary documentation, and ensuring that all communications are clear and compliant.
- Product Lifecycle Management: RA professionals oversee compliance throughout the entire product lifecycle. This includes post-marketing activities such as reporting adverse events, ensuring labeling and advertising comply with regulations, and managing product recalls if needed.
- Strategic Advisory Role: Regulatory professionals provide strategic input during the product development process, advising on regulatory pathways, timelines, and requirements, which can have a significant impact on the success of the product launch.
- International Regulations: For companies operating globally, RA professionals ensure compliance with multiple regulatory frameworks in different regions. This requires knowledge of local regulations, as well as international standards, such as those set by the International Council for Harmonisation (ICH) or the World Health Organization (WHO).
- Ethics and Risk Management: These professionals ensure that ethical considerations, patient safety, and risk management are prioritized in the development and marketing of products. They also help in handling crisis management situations such as safety alerts, product modifications, or adverse event reporting.
- Attention to Detail: RA professionals must be highly detail-oriented as their work involves reviewing complex documentation and ensuring compliance with precise regulatory guidelines.
- Understanding of Science and Law: A strong background in both scientific principles (biology, chemistry, engineering) and legal/regulatory frameworks is essential.
- Project Management: Managing multiple submissions and interacting with various internal teams require strong project management skills.
- Communication Skills: As they interact with both internal teams and regulatory agencies, they need excellent written and verbal communication skills.
- Entry-Level: Often begins as a regulatory affairs assistant or associate, handling documentation and assisting with submissions.
- Mid-Level: As they gain experience, professionals may become regulatory specialists or managers, overseeing submissions and providing more strategic regulatory guidance.
- Senior-Level: Senior positions like directors or vice presidents of regulatory affairs involve overseeing entire RA departments, guiding corporate strategy, and ensuring that all product lines comply with evolving regulations.
- Education: Most professionals in regulatory affairs have a background in science, pharmacy, engineering, or law. Advanced degrees such as Master’s in Regulatory Affairs, Public Health, or Pharmaceutical Sciences can be beneficial.
- Certifications: Certifications such as the Regulatory Affairs Certification (RAC) offered by the Regulatory Affairs Professionals Society (RAPS) can enhance credibility and career prospects in the field.
- In Regulatory Affairs, receiving a Warning Letter or a Form 483 from the FDA presents significant challenges, as both indicate compliance issues and potential risks to product quality, safety, and efficacy. The key challenges regulatory professionals face related to these notices include:
- Complex Regulatory Requirements: The FDA enforces a broad range of regulations that evolve continuously. Keeping up with changes in Good Manufacturing Practices (GMPs), labeling requirements, and clinical trial protocols can be difficult. Misinterpretation or non-compliance often leads to FDA observations in a Form 483, which can escalate to a Warning Letter.
- Inadequate Quality Systems: A robust quality management system is critical for preventing FDA citations. Deficiencies in documentation, training, internal audits, and Corrective and Preventive Action (CAPA) processes are common reasons for Form 483 observations. Establishing and maintaining an effective system requires ongoing commitment and resources, which can be a challenge for companies of varying sizes.
- Data Integrity Issues: Data integrity violations are a growing concern, particularly with the increasing use of electronic systems. Inconsistent, incomplete, or falsified data can trigger a Form 483 and, if not addressed properly, result in a Warning Letter. Regulatory professionals must ensure that their organization’s data practices are secure, transparent, and compliant with FDA standards.
- Resource Constraints: Small and mid-sized companies often face resource limitations, making it difficult to allocate the necessary expertise and budget to ensure full compliance with FDA regulations. This challenge can lead to insufficient internal audits, poorly documented processes, and inadequate training programs, increasing the risk of regulatory violations.
- Supply Chain Oversight: Ensuring compliance extends beyond internal operations to include third-party suppliers and contractors. Regulatory professionals must implement strong oversight mechanisms for their supply chain, as any gaps in compliance among suppliers can lead to FDA citations during inspections. Managing this effectively is often complex and resource-intensive.
- Delayed Response to Observations: Once a Form 483 is issued, companies must respond promptly and comprehensively to avoid escalation to a Warning Letter. Delays or incomplete responses can create further complications, including heightened FDA scrutiny and potential enforcement actions. Developing a rapid and effective response plan is often a logistical and strategic challenge.
- Lack of Proactive Regulatory Strategy: Companies that adopt a reactive approach to regulatory compliance—responding to issues only when they arise—are at greater risk of receiving FDA observations. A proactive strategy, including routine self-inspections, risk assessments, and continuous improvement initiatives, is essential to prevent regulatory citations. However, implementing such a strategy requires foresight and planning, which can be challenging in fast-paced or resource-limited environments.
- Training and Awareness Gaps: Ensuring that all staff, from production to management, are fully aware of FDA requirements and trained accordingly is essential. Gaps in knowledge or inadequate training programs often contribute to non-compliance issues that trigger Form 483 observations. Building a culture of compliance is difficult, especially in organizations where regulatory awareness is low.
These challenges underscore the importance of having a comprehensive and adaptive compliance program to prevent and respond to FDA warning letters and Form 483s.
Most Often Purchased By Regulatory Affairs Professionals
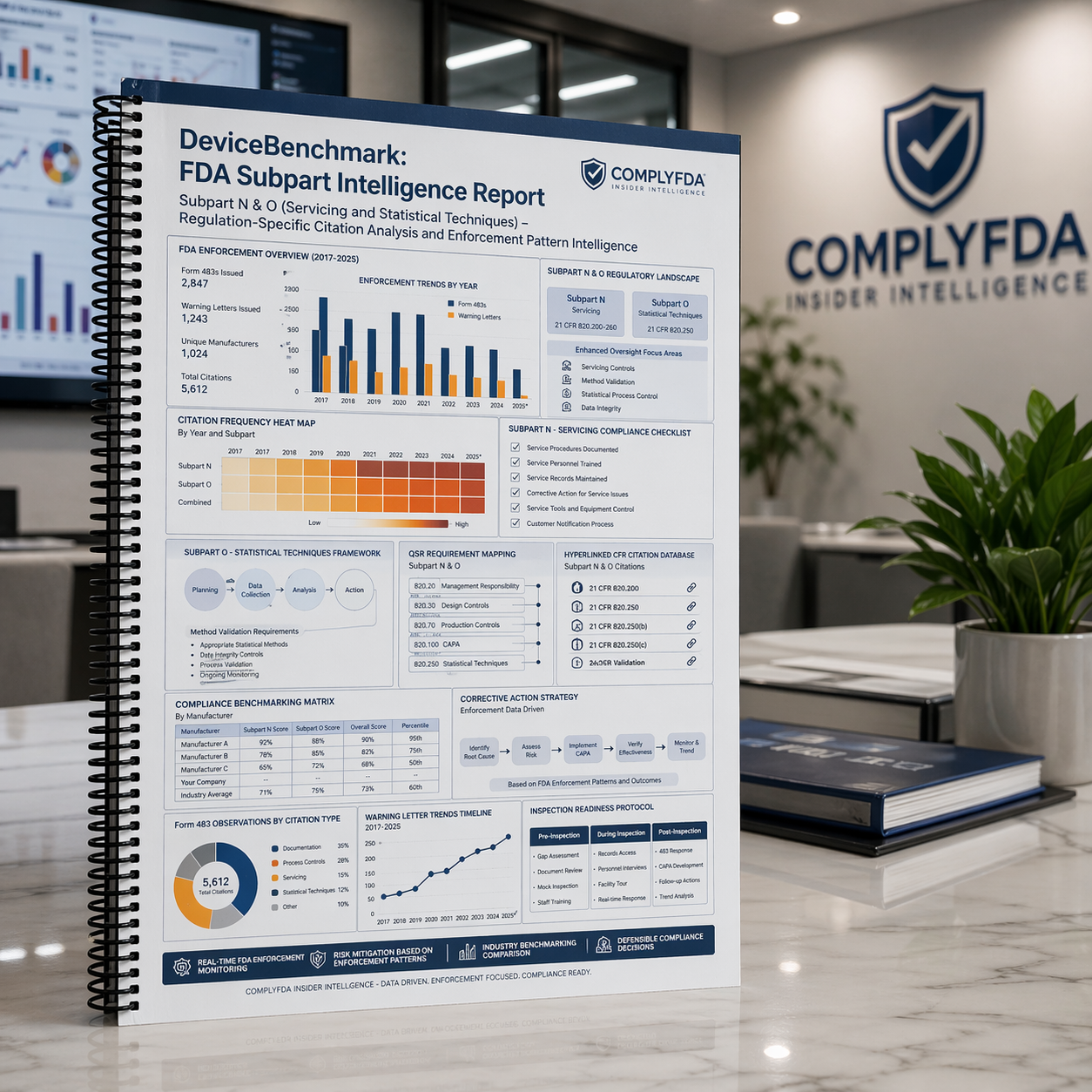
DeviceBenchmark: FDA Subpart Intelligence Report – Subpart N & O (Servicing and Statistical Techniques)
Original price was: $295.00.$250.00Current price is: $250.00.Regulatory Affairs Professionals are the backbone of ensuring product safety and compliance, but with the constant changes in cGMP regulations and the increasing pressure of FDA inspections, the stakes have never been higher. One slip can lead to a Form 483 or a Warning Letter—costing time, money, and potentially derailing your product’s path to market.
ComplyFDA’s Intelligent Insights is the tool you need to stay ahead of these challenges. Don’t wait for an FDA inspection to catch you off guard. Our cutting-edge platform delivers real-time, actionable data tailored to your specific regulatory needs. With features like advanced risk assessments, FDA inspection trend tracking, and automated compliance reports, you’ll know where the risks are before they become costly problems.
Imagine having instant access to the latest FDA inspection trends, so you can proactively address potential issues before they escalate. Avoid the nightmare of a regulatory violation and the ripple effect it can have on your business. Stay audit-ready, reduce compliance risks, and ensure your products consistently meet the highest cGMP standards.
Don’t just react to compliance issues—prevent them. Subscribe to ComplyFDA today, and give yourself the competitive edge to navigate the complexities of regulatory affairs with confidence. Stay compliant, stay competitive, and stay ahead of the FDA.
